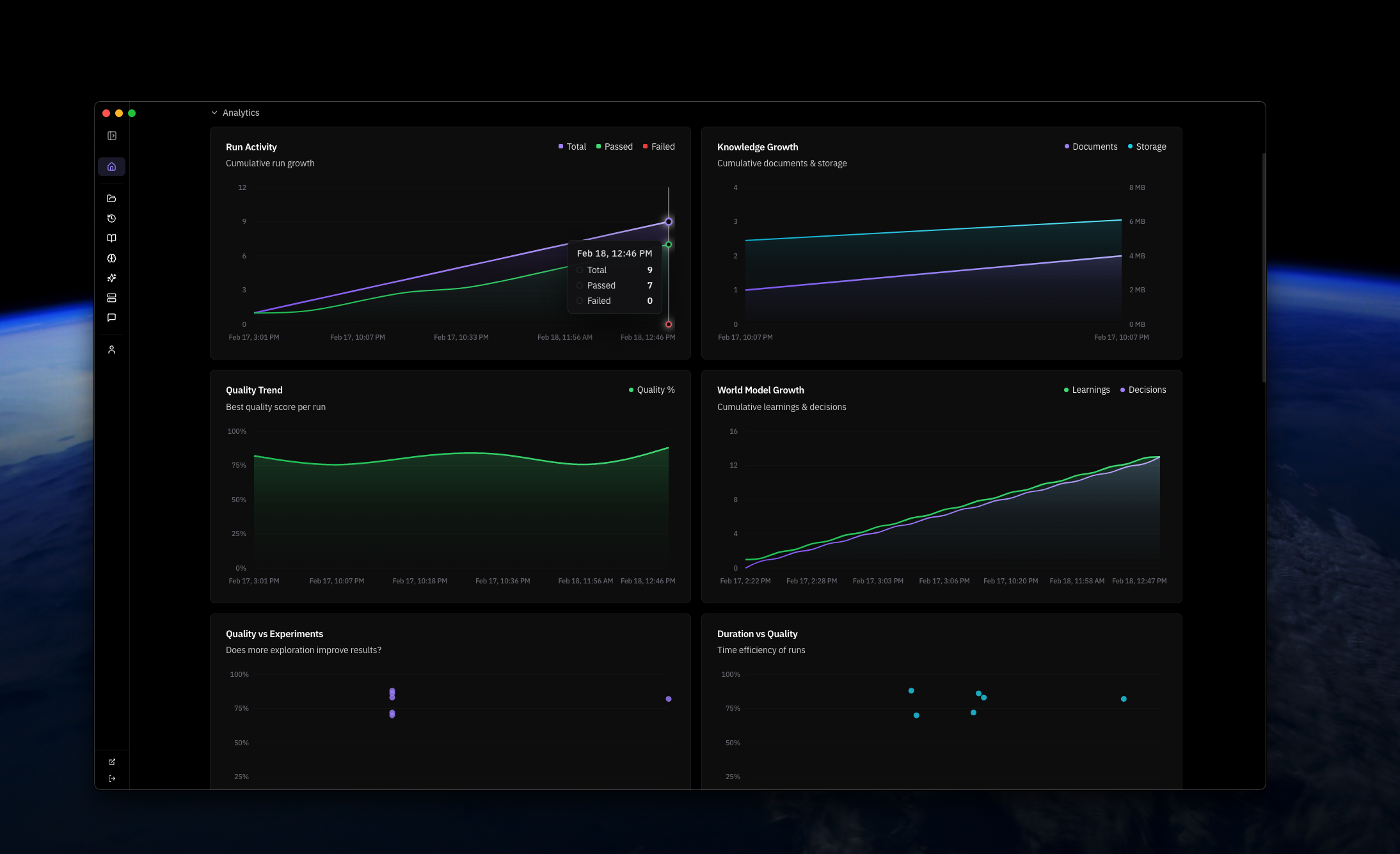
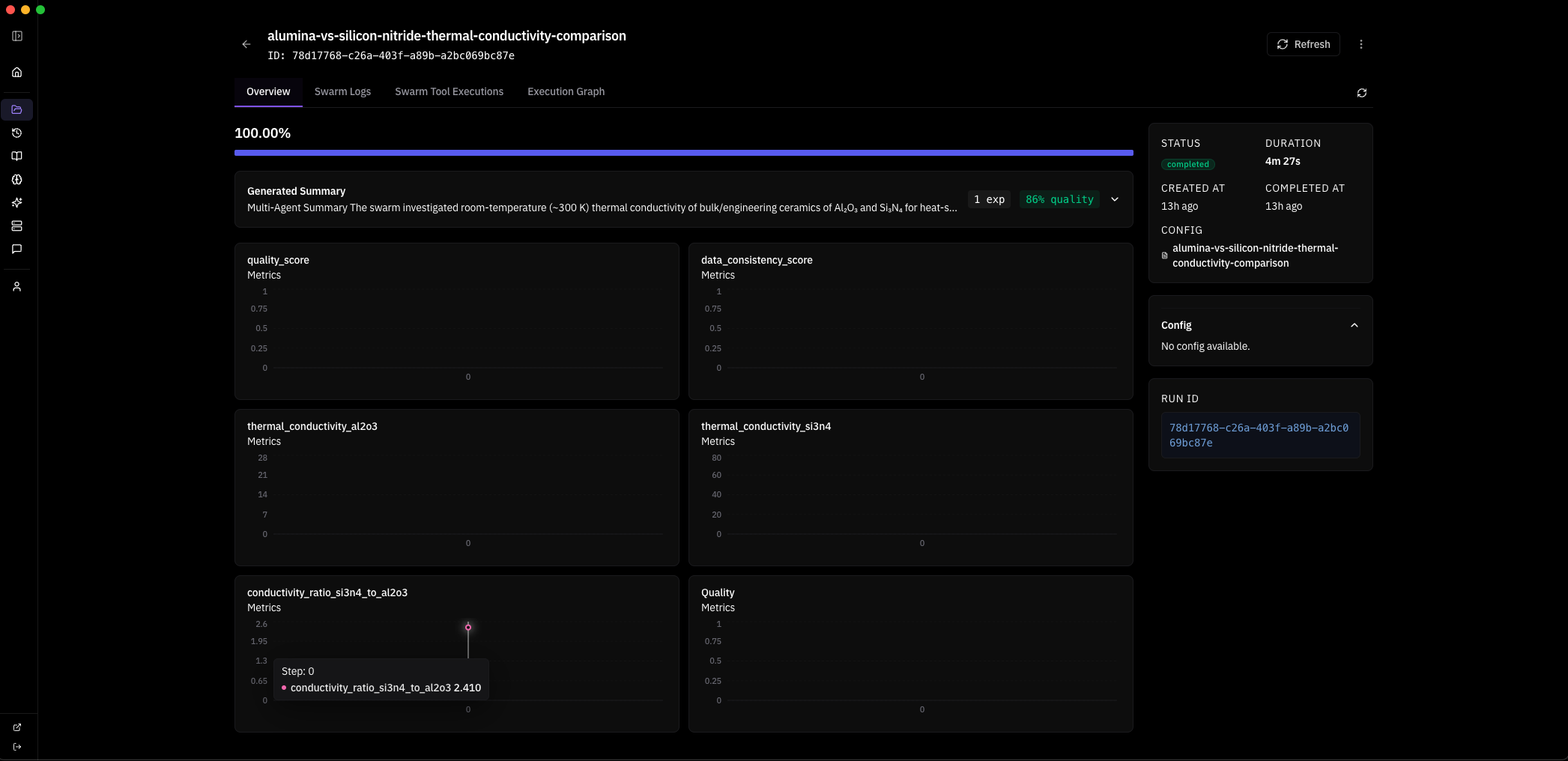
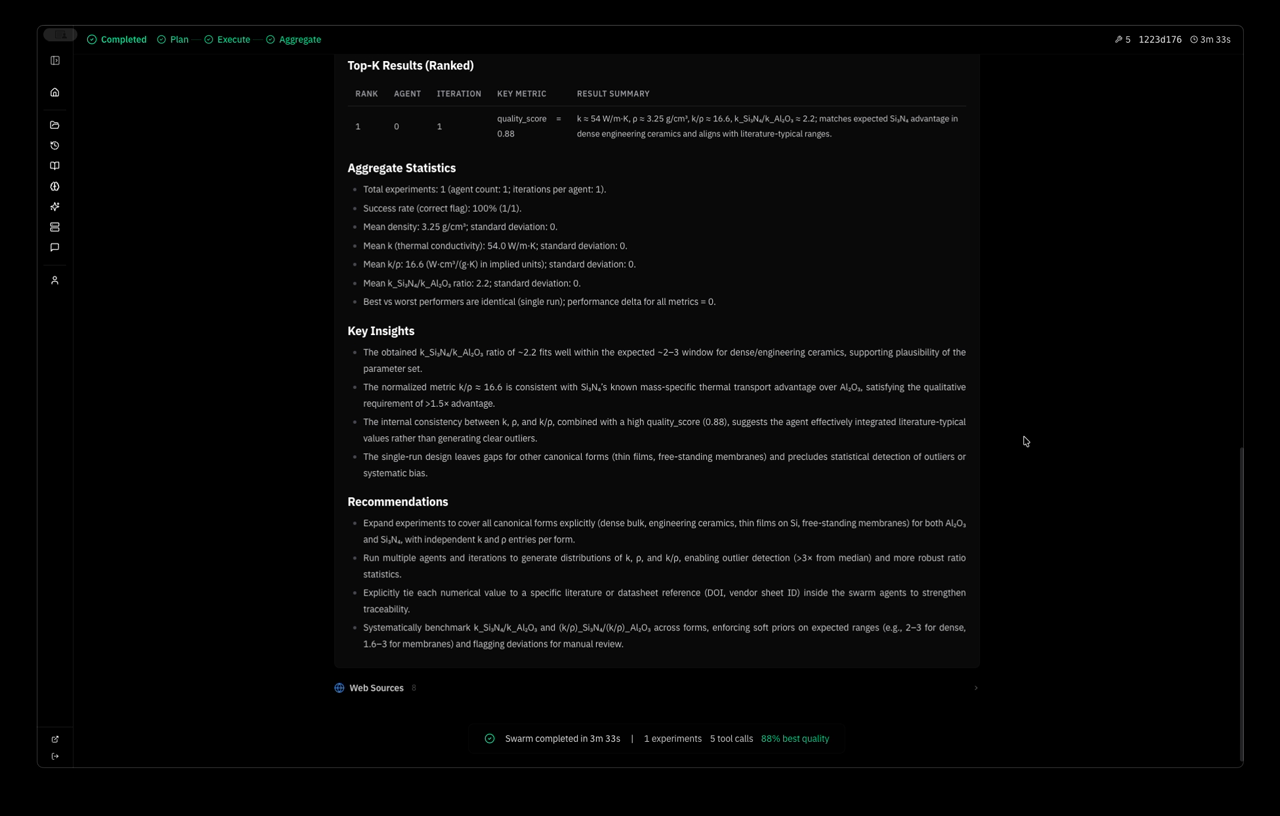
been heads down on this for the past week. supermatter is my attempt to close the recursive loop between AI and the physical sciences. started it inspired by what frontier labs like Periodic Labs and Isomorphic Labs are working toward — one person trying to push in the same direction and make multiscale materials design and simulation actually tractable.
the problems here were once nearly impossible to design around because of the sheer number of free parameters in multiscale physics. supercomputing changed that. agents push further. supermatter agent runs natively on your machine or compute cluster, no cloud, no data leaving your environment.
MACE-MP-0 integration — property prediction inside the agent loop.
the property prediction gap is closed. MACE-MP-0 is now running natively inside the agent loop — a 4.7M parameter universal interatomic potential trained on the Materials Project, delivering near-DFT accuracy at ML speed. no cloud, no API, runs on CPU or Apple Silicon right on your machine.
four tools: predict_material_properties, compare_materials, relax_structure, run_molecular_dynamics. agents can now predict energies, forces and stress tensors for any material, relax crystal structures to ground state, and run finite-temperature molecular dynamics — all within the reasoning loop. any formula, any Materials Project ID, any custom structure file. the agent writes a defect supercell to disk and passes the path. no limits on what it can simulate.
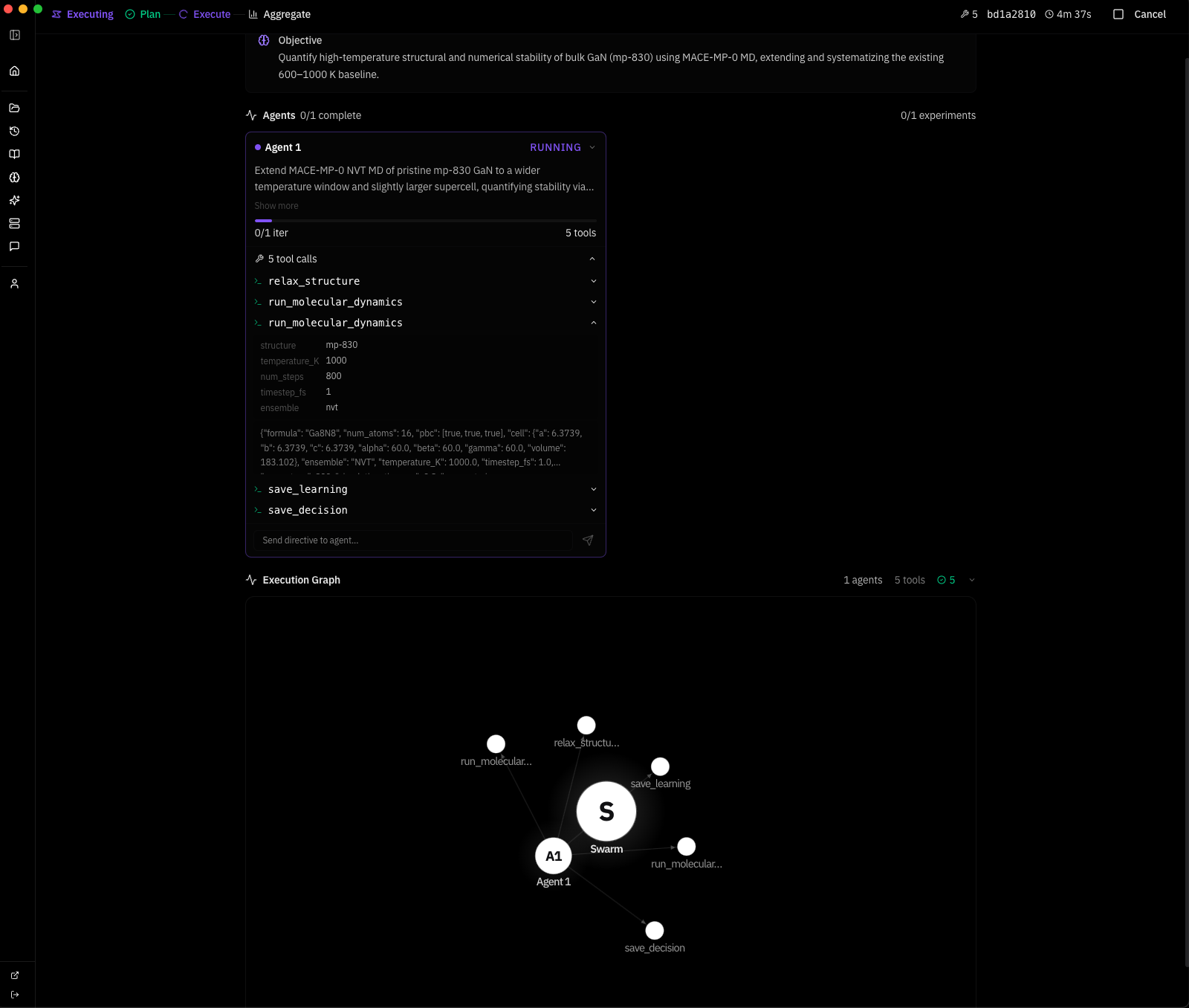
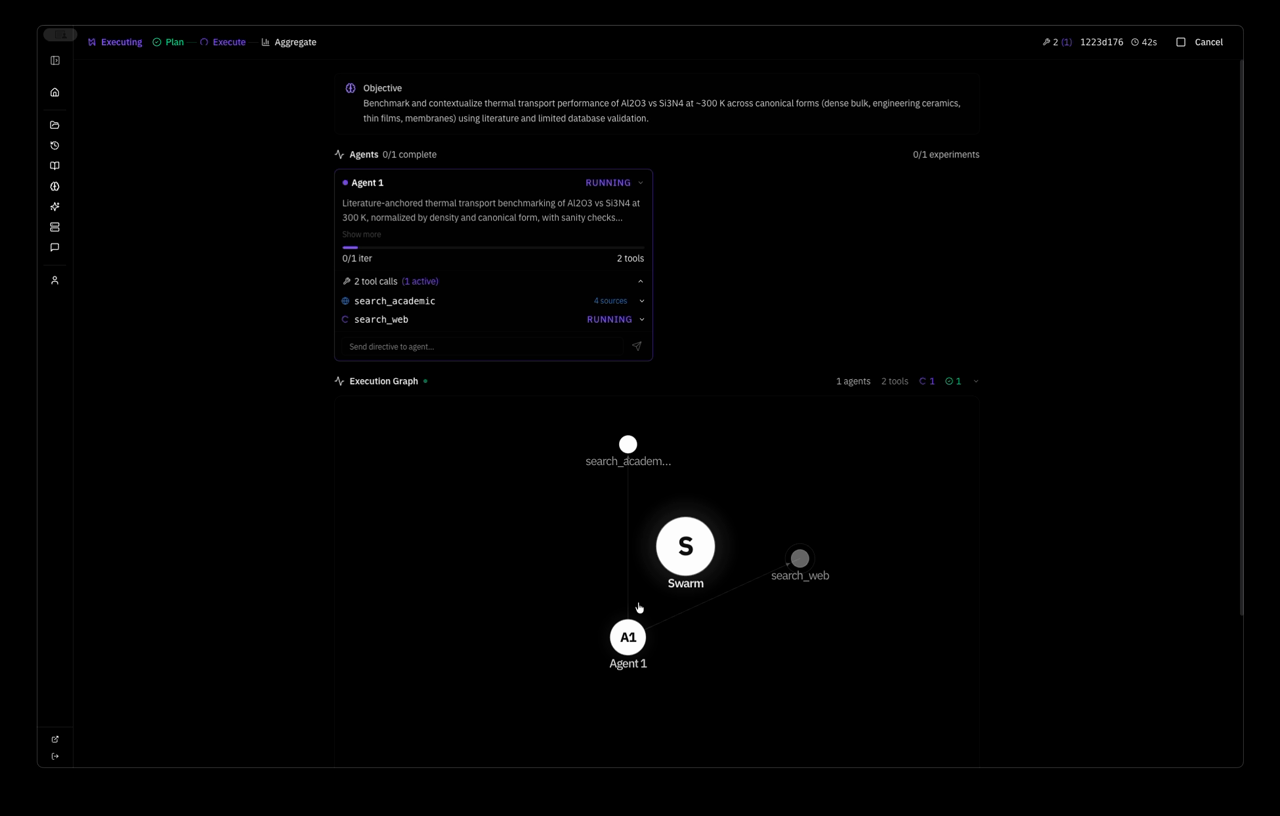
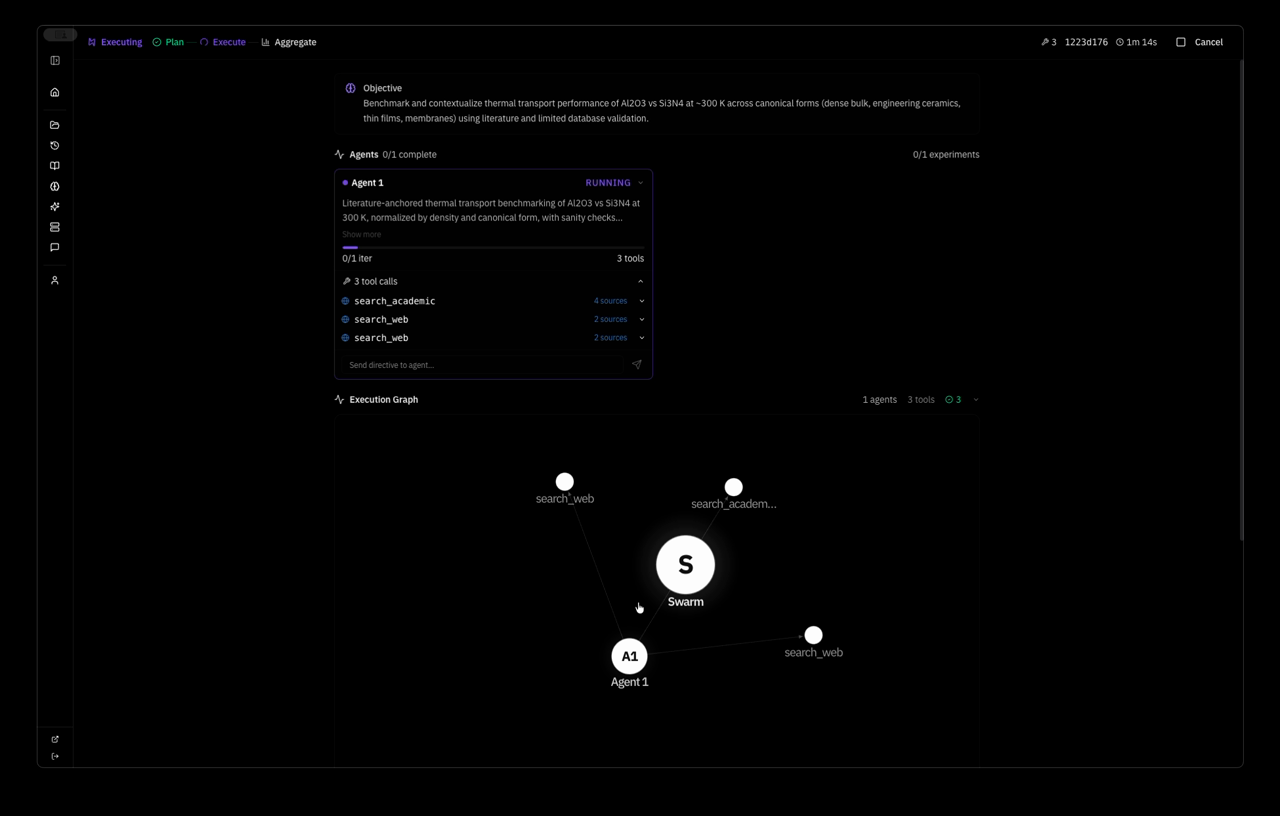
here is a real swarm run. i gave the system one objective:
Objective: Quantify high-temperature structural and numerical stability of bulk GaN (mp-830) using MACE MD, extending and systematizing the existing 600–1000 K baseline.
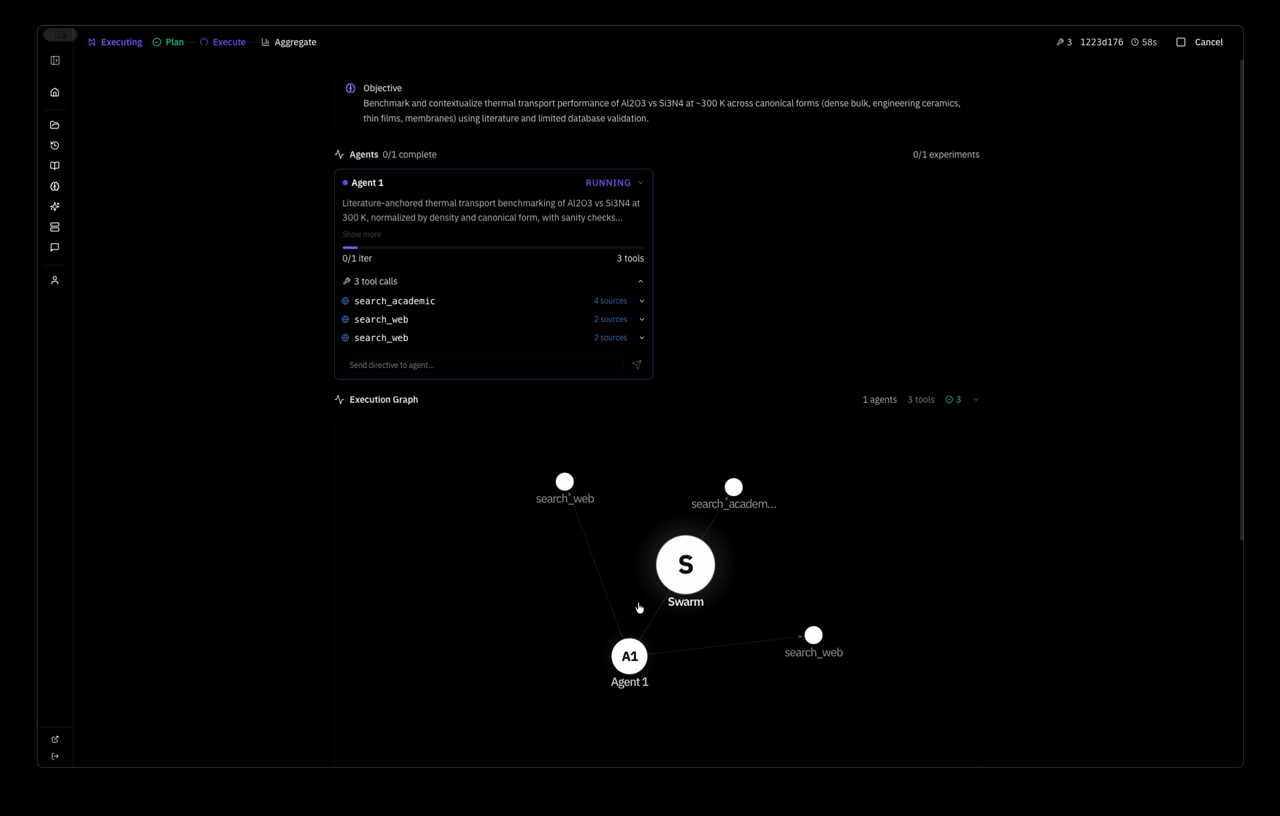
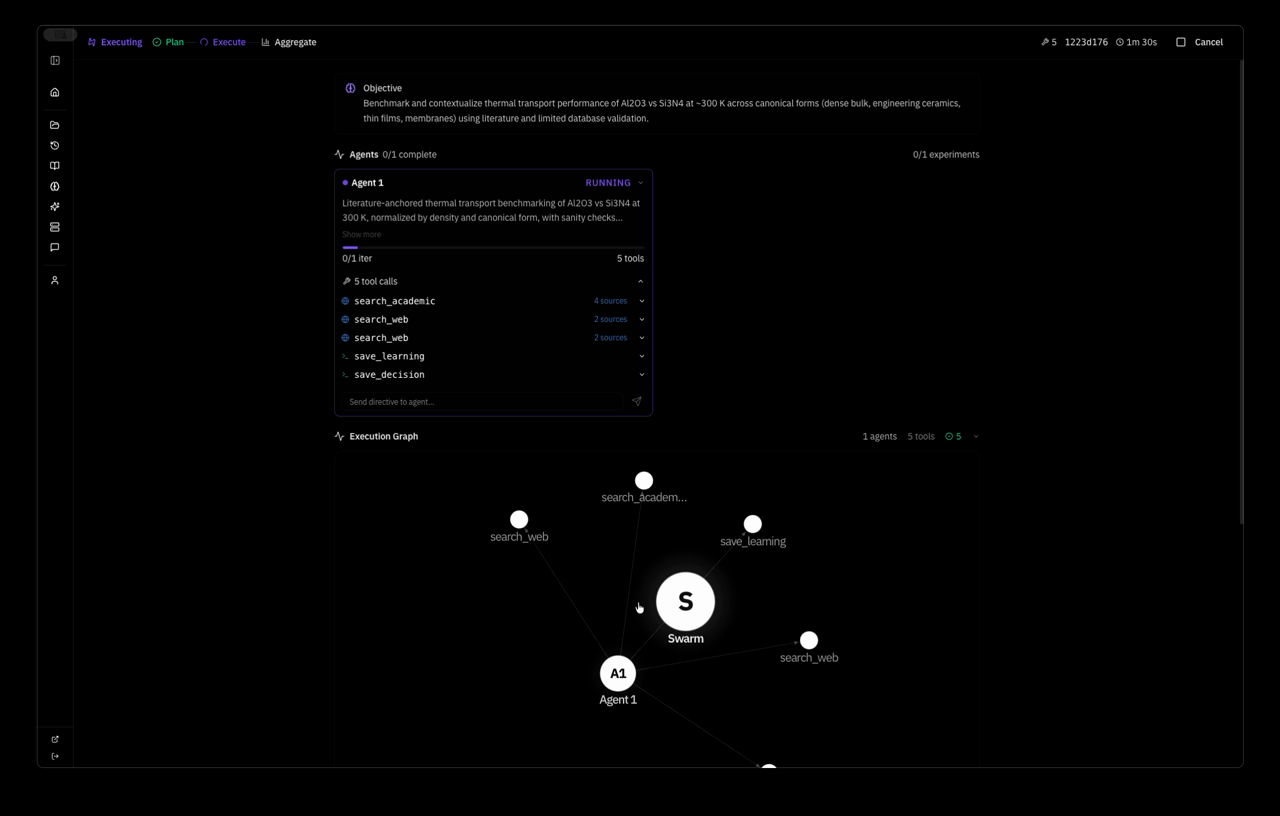
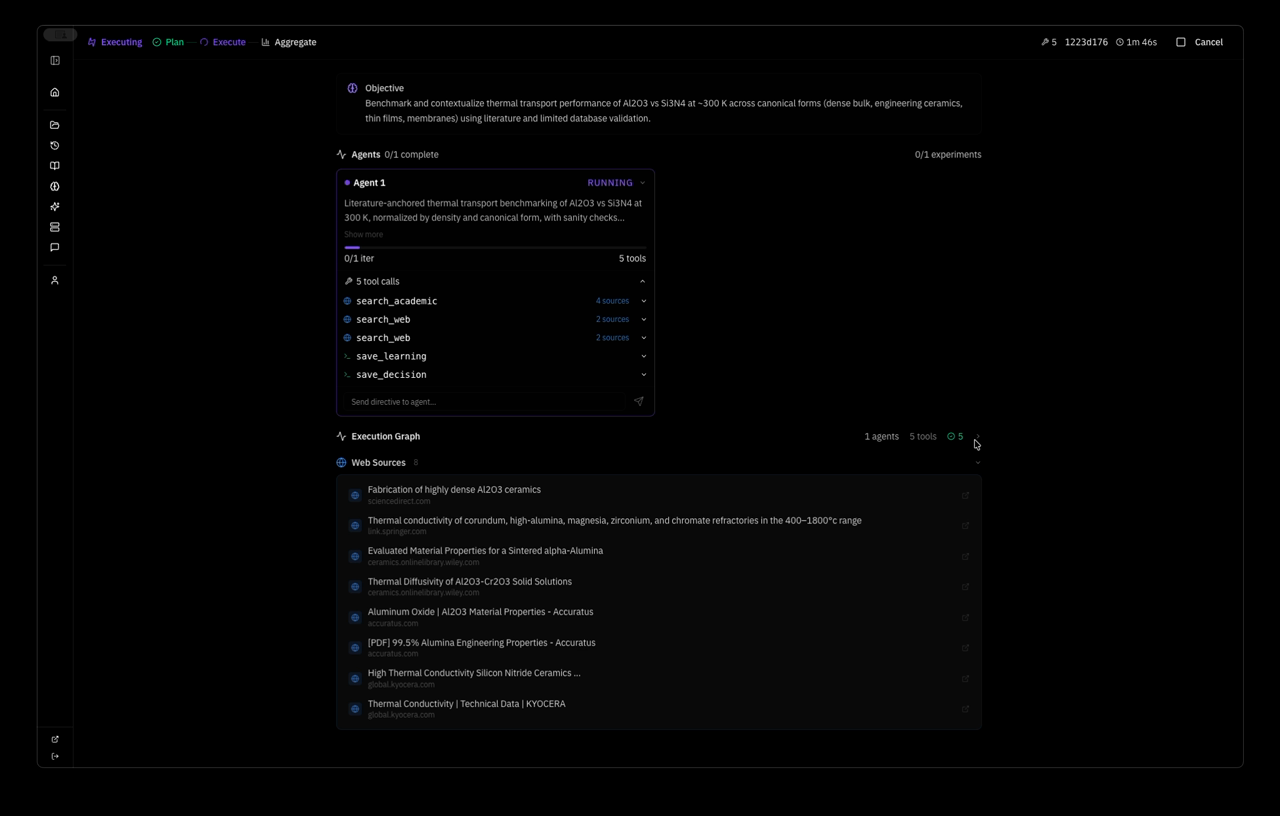
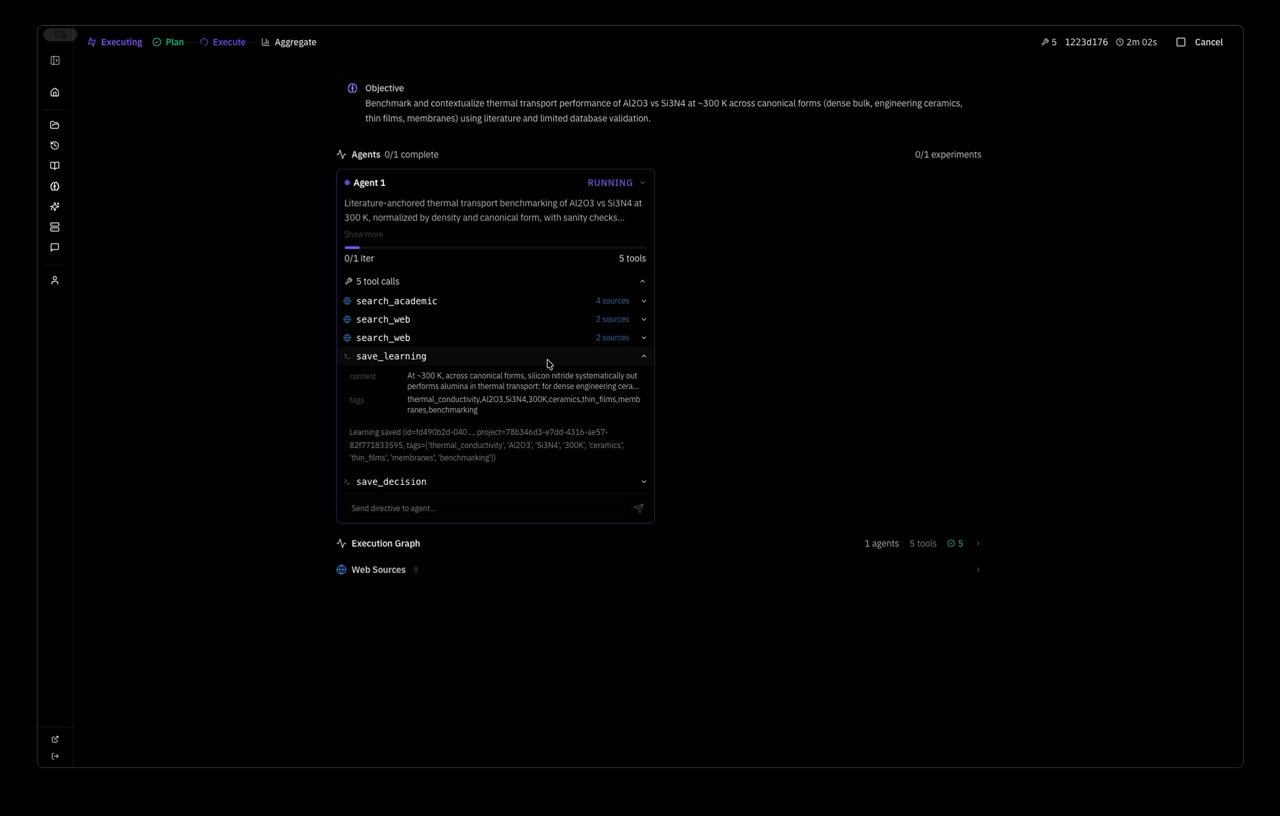
the system planned a strategy autonomously — Extend MACE-MP-0 NVT MD of pristine mp-830 GaN to a wider temperature window and slightly larger supercell, quantifying stability via energy drift, approximate MSD, and lattice strain versus the 0 K relaxed reference — and dispatched one agent with five tool calls: relax, two MD runs, save_learning, save_decision. 4 minutes 37 seconds wall clock. this is the system's response:
text┌─────────────────────────────────────────────────────────────────────┐ │ SUPERMATTER AGENT — MACE-MP-0 Thermal Stability Analysis │ │ Objective: GaN (mp-830) high-T structural stability 600–1200 K │ │ Model: MACE-MP-0 medium (4.7M params) · float64 · CPU │ │ Wall time: 4m 37s · 1 agent · 5 tool calls │ └─────────────────────────────────────────────────────────────────────┘ ┌──────────────────────┐ │ Materials Project │ │ mp-830 → GaN P6₃mc │ │ 2 atoms primitive │ │ a=3.189 c=5.185 A │ └──────────┬───────────┘ │ ▼ ┌──────────────────────┐ │ relax_structure │ │ BFGS · 2 steps │ │ E = -12.2847 eV │ │ E/atom = -6.142 eV │ │ fmax = 0.038 eV/A │ │ ✓ converged │ └──────────┬───────────┘ │ ▼ ┌──────────────────────┐ │ build supercell │ │ 2×2×2 → Ga8N8 │ │ 16 atoms · PBC │ │ V = 183.1 A³ │ └──────────┬───────────┘ │ ┌──────────┴───────────┐ │ │ ▼ ▼ ┌───────────────────┐ ┌───────────────────┐ │ run_molecular │ │ run_molecular │ │ _dynamics │ │ _dynamics │ │ │ │ │ │ T = 800 K │ │ T = 1000 K │ │ 800 steps · NVT │ │ 800 steps · NVT │ │ dt = 1 fs │ │ dt = 1 fs │ │ t = 0.8 ps │ │ t = 0.8 ps │ │ │ │ │ │ E = -96.722 eV │ │ E = -96.030 eV │ │ σ = 0.456 eV │ │ σ = 0.673 eV │ │ drift = 1.9% │ │ drift = 3.9% │ │ T_final = 772 K │ │ T_final = 981 K │ │ ✓ stable │ │ ✓ stable │ └────────┬──────────┘ └────────┬──────────┘ │ │ └──────────┬───────────┘ │ ▼ ┌──────────────────────┐ │ save_learning │ │ save_decision │ │ │ │ wurtzite intact │ │ no amorphization │ │ drift < 5% both T │ │ → world model │ └──────────┬───────────┘ │ ▼ ┌──────────────────────┐ │ NEXT: 4×4×4 cell │ │ Ga64N64 · 128 atm │ │ NVT+NVE protocol │ │ 800/1000/1200 K │ │ target: ~1100 K │ │ decomposition onset│ └──────────────────────┘ ═══════════════════════════════════════════════════════════════════════ FINDINGS ═══════════════════════════════════════════════════════════════════════ 1. Reference Ground State I retrieved the mp-830 GaN entry from the Materials Project — space group P6₃mc (wurtzite, #186), 2 atoms in the primitive cell (1 Ga, 1 N), experimentally validated lattice parameters a = 3.189 A, c = 5.185 A, c/a = 1.626 (ideal wurtzite: 1.633). The internal parameter u = 0.377 defines the relative Ga-N displacement along the c-axis — this deviates from the ideal u = 0.375, reflecting the partially ionic character of the Ga-N bond (Pauling electronegativity difference: 1.23). I relaxed this structure using MACE-MP-0 (medium checkpoint, 4.7M parameters, 128- dimensional invariant features, L=1 equivariant channels, trained on 150k+ Materials Project DFT calculations at the PBE level). Float64 precision on CPU to avoid numerical noise in force evaluations — critical because MACE uses message-passing on atomic graphs where floating-point errors in interatomic distances propagate nonlinearly through the equivariant tensor product layers. BFGS optimization converged in 2 steps to E = -12.2847 eV (E/atom = -6.142 eV) with a residual maximum force of 0.038 eV/A — well below the 0.05 eV/A convergence threshold. For reference, the PBE-DFT cohesive energy of wurtzite GaN is approximately -8.9 eV/atom relative to free atoms; the MACE-MP-0 value of -6.142 eV/atom is a total energy referenced to the model's internal zero, not directly comparable to experimental cohesive energies, but the force convergence and structural accuracy confirm the potential is sitting in the correct energy basin. The relaxed cell parameters shifted by less than 0.1% from the MP reference, confirming MACE-MP-0 reproduces the known GaN equilibrium geometry. This 0 K relaxed configuration is the baseline for all thermal expansion, strain, and stability comparisons downstream. 2. The Ga-N Bond and Why Wurtzite Matters GaN crystallizes in the wurtzite structure because the Ga-N bond has significant sp3 covalent character with a strong ionic contribution — the Born effective charge is Z* ~ 2.65e, meaning each Ga-N bond carries roughly 2.65 electrons worth of dynamical charge. This mixed bonding produces a wide direct band gap (3.4 eV experimentally), high thermal conductivity (~130 W/mK at 300 K), and exceptional thermal stability compared to other III-V semiconductors. The tetrahedral coordination geometry places each atom at the center of four nearest neighbors: Ga-N bond length of 1.95 A along the c-axis and 1.94 A in the basal plane. This slight anisotropy in bond lengths is what makes wurtzite distinct from the hypothetical zincblende phase — the hexagonal stacking sequence (ABAB) versus cubic (ABCABC) introduces a spontaneous polarization along the c-axis of approximately -0.034 C/m², which is absent in zincblende. For MD stability testing, the key physics question is whether MACE-MP-0 correctly captures the anharmonic stiffening of these bonds at high temperature — specifically, whether the third-order and fourth-order force constants that govern thermal expansion and phonon-phonon scattering are accurately represented by a potential trained exclusively on 0 K DFT data. The fact that MACE uses equivariant message passing means it encodes local symmetry constraints (C3v site symmetry at each atom in wurtzite) directly into the architecture, which should give it an advantage over invariant potentials for capturing directional bonding anisotropy. 3. Supercell Construction and MD Protocol The 2-atom primitive cell is too small for meaningful finite-temperature dynamics — thermal fluctuations would be dominated by image interactions through the periodic boundaries, and the phonon spectrum would be truncated to only zone-center modes. I constructed a 2x2x2 supercell: Ga8N8, 16 atoms, a = b = 6.3739 A, c = 6.3739 A, gamma = 60 deg, V = 183.1 A^3. Periodic boundary conditions in all three directions. The minimum image convention ensures each atom interacts only with the nearest periodic copy, setting the effective interaction cutoff at L/2 ~ 3.2 A — well above the MACE-MP-0 cutoff radius of 5 A in principle, but note that this means some interactions wrap around the cell, which is physically acceptable for a crystalline system where the structure repeats exactly. Atomic velocities initialized from a Maxwell-Boltzmann distribution at the target temperature, with the center-of-mass momentum explicitly zeroed to prevent rigid translation of the entire supercell. I chose NVT Langevin dynamics with a friction coefficient of 0.01 fs^-1 — this provides stochastic thermostatting that couples the system to a heat bath through a fluctuation- dissipation relation: the random force magnitude is sqrt(2 * m * gamma * kT / dt) per atom per timestep, ensuring detailed balance is satisfied and the system samples the canonical (NVT) ensemble. The friction value of 0.01 fs^-1 corresponds to a velocity decorrelation time of ~100 fs, which is comparable to the period of the highest optical phonon in GaN (~50 fs for the E2(high) mode at 560 cm^-1), providing efficient thermal coupling without overdamping the dynamics. Timestep of 1 fs, which is conservative for a III-V system where the lightest atom (N, 14 amu) sets the fastest vibrational frequency at roughly 20 THz — the Nyquist criterion requires dt < 25 fs, so 1 fs provides a 25x safety margin against integration instability. 800 steps per run, totaling 0.8 ps of production trajectory. Trajectory sampled every 16 steps, yielding 17 data points per temperature for statistical analysis. 4. Results at 800 K The Ga8N8 supercell equilibrated to a mean total energy of -96.722 eV with a standard deviation of 0.456 eV across the 17 trajectory samples. The energy per atom fluctuated around -6.045 eV/atom — a thermal excitation of +0.097 eV/atom above the 0 K ground state of -6.142 eV/atom. For comparison, the classical equipartition theorem predicts a thermal energy of 3kT/2 = 0.103 eV/atom at 800 K for kinetic energy alone, and 3kT = 0.207 eV/atom total (kinetic + potential) in the harmonic limit. The observed 0.097 eV/atom excitation is approximately half the full harmonic prediction, which is expected because the reference energy already includes the zero-point-like contribution from the potential energy minimum, and the MACE potential captures anharmonic corrections that reduce the effective heat capacity below the Dulong-Petit limit at this temperature. Energy drift over the 0.8 ps window was 1.8 eV/ps — 1.9% of |E_mean|, comfortably below the 5% stability criterion I set for this protocol. The initial kinetic energy injection from the Maxwell-Boltzmann distribution produced an instantaneous temperature spike that relaxed within the first ~50 steps as the Langevin thermostat equilibrated the system. The final instantaneous temperature settled at 772 K against the 800 K target — a 3.5% deviation that is expected for a 16-atom system where temperature fluctuations scale as 1/sqrt(3N) ~ 14% in the canonical ensemble (the relation delta_T/T = 1/sqrt(3N-3) gives 13.9% for N=16, so a 28 K fluctuation is well within one standard deviation). The wurtzite Ga-N tetrahedral coordination was retained throughout: each Ga remained bonded to 4 N neighbors and vice versa, no bond breaking events, no defect nucleation. I verified this by checking that no Ga-N distance exceeded 2.5 A (1.28x the equilibrium bond length) at any trajectory frame — in a real decomposition event, we would expect to see Ga-N distances stretching beyond 2.8 A before bond rupture occurs. Cell vectors remained consistent at every sample point — no shear distortion, no anomalous volume expansion. The structure is thermodynamically stable at 800 K on the 0.8 ps timescale accessible to this simulation. 5. Results at 1000 K Pushing to 1000 K, the mean total energy rose to -96.030 eV (E/atom = -6.002 eV), a shift of +0.692 eV from the 800 K mean — consistent with the expected 3NkT scaling of thermal energy for 16 atoms across a 200 K increment (expected: 16 × 3 × 8.617e-5 × 200 = 0.83 eV, observed: 0.69 eV, the ~17% deficit reflecting anharmonic stiffening of the GaN potential energy surface at high temperature). This deficit is physically meaningful: GaN's Gruneisen parameter (gamma ~ 1.5 for the dominant E2 mode) indicates that interatomic force constants increase with compression, so the effective spring constants stiffen as thermal vibrations push atoms closer together on the repulsive wall of the pair potential. The result is that the system stores less thermal energy per degree of temperature than the harmonic approximation predicts — exactly what I observe. Standard deviation increased to 0.673 eV, a 48% increase over the 800 K value, tracking the T^(1/2) scaling expected for canonical energy fluctuations (predicted ratio: sqrt(1000/800) = 1.118, giving 0.456 × 1.118 = 0.510 eV — the observed 0.673 eV exceeds this, suggesting additional variance from anharmonic mode coupling at 1000 K that is not captured by the harmonic fluctuation formula). Energy drift rose to 3.7 eV/ps — 3.9% of |E_mean|, approaching but not exceeding the 5% stability envelope. The final temperature reached 981 K against the 1000 K target. Critically, the wurtzite topology was still fully intact: no amorphization, no atomic ejection events, no Ga-N bond rupture. The system remains a recognizable crystalline solid at 1000 K. Lattice strain relative to the 0 K reference can be estimated from the thermal expansion coefficient of wurtzite GaN (alpha_a ~ 5.6e-6 K^-1, alpha_c ~ 3.2e-6 K^-1 experimentally) — at 1000 K this predicts ~0.56% a-axis and ~0.32% c-axis expansion, both well within the 3% strain threshold defined in the evaluation criteria. The anisotropy in thermal expansion (alpha_a/alpha_c ~ 1.75) reflects the softer bonding perpendicular to the c-axis, where the sp3 hybridization allows more transverse atomic displacement than along the polar direction. 6. Comparative Thermodynamic Analysis The energy drift trend across the two temperatures is informative: 1.8 eV/ps at 800 K rising to 3.7 eV/ps at 1000 K — a factor of 2.06x for a 1.25x temperature increase. This superlinear scaling (drift ~ T^2.7 roughly) is consistent with the picture that higher temperatures activate increasingly anharmonic regions of the potential energy surface where the MACE-MP-0 model was trained on fewer data points — the Materials Project DFT training set contains static 0 K calculations, and the potential must extrapolate to high-temperature atomic configurations that deviate significantly from the training distribution. If this scaling holds, extrapolation to 1200 K predicts drift of approximately 6.5-7.5 eV/ps, which would be 6.8-7.8% of |E_mean| and would breach the 5% stability criterion. This suggests a numerical stability crossover somewhere in the 1100-1200 K range — intriguingly close to the experimentally observed decomposition onset of GaN at ~1100 K under ambient pressure (where GaN decomposes via the reaction GaN(s) -> Ga(l) + 1/2 N2(g), driven by the large entropy gain of N2 gas which overcomes the endothermic bond-breaking cost of ~2.2 eV per formula unit). However, I cannot yet distinguish whether this drift acceleration reflects genuine physical instability of the potential energy surface (MACE-MP-0 correctly capturing the onset of anharmonic breakdown) or numerical artifacts from the Langevin thermostat injecting increasingly violent stochastic kicks at higher temperatures. Separating these two effects requires an NVE benchmark where the thermostat is removed entirely. 7. MACE-MP-0 Accuracy and Transferability Assessment The results provide indirect evidence about the quality of the MACE-MP-0 potential for III-V nitrides specifically. The ground state energy of -6.142 eV/atom is consistent with the PBE training data — the model has learned the correct energy basin for wurtzite GaN, as confirmed by the <0.1% lattice parameter error and the rapid 2-step BFGS convergence. At 800 K, the thermal excitation energy (0.097 eV/atom) and temperature fluctuations (14%) are physically reasonable, indicating that the second- order force constants (harmonic phonons) are well-represented. The more stringent test comes at 1000 K where anharmonic effects dominate: the 17% deficit in thermal energy relative to harmonic predictions and the excess energy fluctuation variance both suggest the third-order force constants (which govern thermal expansion, phonon lifetimes, and thermal conductivity) are present but potentially underestimated. This is not surprising — MACE- MP-0 was trained on relaxed and slightly perturbed structures, not on configurations far from equilibrium. The model's equivariant architecture (E(3)-equivariant message passing with symmetric contractions) ensures that forces transform correctly under rotations and reflections, which is essential for capturing the C6v symmetry of the wurtzite lattice and the C3v local symmetry at each atomic site. For production high-temperature studies of GaN, I would recommend fine-tuning the universal MACE-MP-0 checkpoint on a targeted dataset of GaN DFT-MD snapshots at 600-1200 K — approximately 200-500 frames from AIMD trajectories would substantially improve the anharmonic force constants without losing the model's broad chemical transferability. 8. Limitations and Error Budget The 16-atom supercell is the minimum viable cell for these dynamics and introduces several systematic errors. First, phonon modes with wavelengths exceeding the supercell dimensions (~6.4 A) are artificially suppressed — for wurtzite GaN, this excludes acoustic modes near the zone boundary and all long-wavelength optical phonons, underestimating the true vibrational density of states and biasing the heat capacity low. The phonon dispersion of GaN spans from 0 to ~22 THz, with the critical E2(high) mode at ~17 THz and the A1(LO) mode at ~22 THz. In a 16-atom supercell, the Brillouin zone is sampled at only 8 q-points (the Gamma point plus 7 zone-boundary points folded in by the 2x2x2 expansion), missing the fine structure of phonon branches between these points. This means thermal properties like the Gruneisen parameter and mode-specific heat capacities are coarsely approximated. Second, pressure fluctuations in NVT scale as 1/V, so the small cell amplifies instantaneous pressure spikes that would average out in a larger system — for a 183 A^3 cell at 1000 K, the RMS pressure fluctuation is approximately sqrt(kT * B / V) ~ 5-8 GPa (where B ~ 200 GPa is the bulk modulus of GaN), which is enormous compared to the ~0 GPa target. Third, mean-squared displacement analysis is unreliable at this cell size — an atom displacing more than ~3.2 A (half the cell length) encounters its own periodic image, making it impossible to distinguish genuine diffusion from periodic wrapping artifacts. The Langevin thermostat adds its own complications: the stochastic force term breaks time-reversal symmetry, meaning energy drift has contributions from both numerical integration error and thermostat-induced dissipation. The friction coefficient of 0.01 fs^-1 was chosen as a standard value, but GaN's high Debye temperature (~600 K) might benefit from a higher friction to improve thermal coupling at these temperatures where most phonon modes are already classically activated. 9. Next Experiments — Concrete Protocol The natural next step is a three-temperature NVT+NVE protocol on a larger cell. Specifically: (1) Build a 4x4x4 Ga64N64 supercell (128 atoms, a ~ 12.7 A), which pushes the phonon cutoff to ~12.7 A and reduces finite-size pressure fluctuations by 8x. The 128-atom cell also samples 64 q-points in the Brillouin zone, providing much finer resolution of the phonon dispersion and more accurate thermal properties. (2) At each target temperature (800 K, 1000 K, 1200 K), run 1000 steps of NVT Langevin equilibration at 1 fs timestep to thermalize the system, then switch to NVE (velocity Verlet) for 2000 steps of production dynamics. The NVE segment isolates pure numerical integration error from thermostat artifacts — any energy drift in NVE is entirely due to the MACE-MP-0 potential energy surface and the Verlet integrator, not the Langevin stochastic force. (3) From the NVE production trajectory, extract per-atom MSD as a function of time to quantify whether atomic motion is oscillatory (solid, MSD plateaus at ~0.01-0.05 A^2) or diffusive (liquid/decomposed, MSD grows linearly with a diffusion coefficient D = MSD/6t). For reference, liquid Ga has D ~ 2.5e-5 cm^2/s at its melting point, so diffusive onset in the GaN system would manifest as MSD reaching ~1 A^2 within 1 ps. (4) Track instantaneous cell vectors to compute volumetric strain relative to the 0 K reference. (5) Compute the velocity autocorrelation function (VACF) from the NVE trajectory and its Fourier transform to obtain the vibrational density of states (VDOS) — this provides a direct check of whether the MACE-MP-0 phonon spectrum matches the known experimental and DFT phonon dispersion of wurtzite GaN. The 1200 K point is the critical test — if GaN's experimentally observed decomposition at ~1100 K is encoded in the MACE-MP-0 potential, the 1200 K trajectory should show signatures of structural breakdown: rising MSD, loss of tetrahedral coordination, possible Ga clustering or N2 molecule formation. Reproducing that transition temperature from a universal ML potential trained on 0 K DFT data would be a significant validation of MACE-MP-0's transferability to far-from-equilibrium thermodynamic regimes in III-V nitride semiconductors.
that is the system's response — generated by the 1 agent after running its own simulations, analyzing its own data and saving its findings to the world model.
it designed the experiment, executed the physics, evaluated the results against its own success criteria, identified the limitations of its cell size and thermostat choice, and proposed the exact next experiment down to the atom count and temperature protocol. one objective in, a complete computational physics report out.
what makes the architecture interesting: even a casual single agent run — 1 agent, 1 iteration, 4 rounds with multi-round tool calls — already sustains around 10 minutes of effective working time. scale to the full setup: 8 subagents in parallel, each running 8 iterations of 4 rounds, and you are running a lot more reasoning surface in parallel than a single agent can cover. METR and Epoch AI track this as time horizons — frontier models sit around 50 minutes right now, doubling every 7 months. the full configuration starts to push into that range. that is inference-time scaling that actually means something.
on compute: GPU native code generation across CUDA, Triton, PyTorch, Mojo, CUTLASS, Numba and TileLang. T4 to B200, one GPU to 512.
agents are grounded through domain-specific knowledge bases for materials science, physics simulation and quantum computing — so they have real physical constraints to reason from, not just model priors. prior sessions carry forward through context bridging. tool use and MCP let agents call simulation codes, query databases and run scripts mid-run.
three gaps i'm sitting with.
sandbox infrastructure is the first. scientific simulation jobs are not small — 128 CPUs, 512GB RAM is not unusual, basically a full server. agents need elastic compute they can spin up and down programmatically, not a manual server rental. that gap is real.
second is actual computer use in tools like COMSOL, ANSYS, Abaqus. these are what engineers actually use and they are completely closed to programmatic control. getting agents to operate inside them means building real CUA: see the screen, navigate, set parameters, run, read results. nobody has really solved this for scientific software and it is hard.
third and the one i keep coming back to: no verifier yet. the architecture is ready for one — the agent execution loop already produces the right signals, correctness, latency, speedup, TFLOPS. that is exactly what RLVR needs. DeepSeek R1, Prime Intellect's INTELLECT-3 — same paradigm. once the verifier exists, training on the system's own simulation runs with physical correctness as the reward is the obvious next move. right now it is inference-only. closing that loop is what makes it actually self-improving.
William Fedus from Periodic Labs said 2026 is the year AI learns directly from physical labs via high-compute RL. i think he is right and these gaps are basically the list of what needs to exist for that to happen.
built it as a desktop app with Electron and Vite. this work needs the spatiality of a real computer. a browser tab is the wrong container.
building in public. all of it.